Anand Lab
Research Publications Group Members BiographyRepair of DNA double-strand breaks by homology-directed repair pathways
The integrity of the DNA in our cells is essential for normal cellular functions. However, DNA frequently encounters many exogenous as well as endogenous DNA-damaging agents and events, which can result in various DNA lesions. While exogenous factors include ionising radiation (IR) from X-rays, ultraviolet (UV) light from the sun and DNA-alkylating and -crosslinking chemicals, errors in normal endogenous processes like DNA replication, transcription, or toxic cellular metabolites like reactive oxygen species (ROS) also regularly 'insult' DNA on a constant basis. To deal with these DNA lesions, cells have evolved multiple DNA repair strategies depending on the type of lesion. In fact, defects in DNA repair pathways underpin various malignancies, accelerated ageing, immunological disorders and severe developmental abnormalities.
DNA double-strand breaks (DSBs) are one of the most dangerous lesions because, unlike lesions affecting single-stranded DNA, cells don’t have intact strands to guide and restore the original sequence when both strands are broken. To deal with this challenge, cells predominantly use one of two DSB repair pathways known as non-homologous end joining (NHEJ) and homologous recombination (HR). In the more error-prone NHEJ, cells simply re-ligate the broken ends together, which can be preceded by end-processing, leading to deletions. Conversely, in HR, cells can either use sister chromatids or homologous chromosomes as templates to copy the sequence for error-free repair of DSBs. Mechanistically, HR is a multi-step process, which involves DNA end resection, homology search, strand invasion, DNA synthesis, strand annealing, and either nucleolytic cleavage or disentanglement of a double-Holliday junction (an HR intermediate), depending on the selected sub-pathway (Figure 1).
Besides NHEJ and HR, additional alternative 'backup' DSB repair pathways exist in cells, which can be employed upon either failure or unavailability of the main repair pathways. One such scenario is depicted in Figure 1. These alternative pathways include single-strand annealing (SSA), alternative-end joining (Alt-EJ), theta-mediated end joining (TMEJ), and break-induced replication (BIR) repair. In contrast to HR, all these alternative repair pathways are error-prone to varying degrees. However, a common feature of all these repair pathways, including HR, is that they use varying degrees of DNA homology to repair DSBs and thus they are collectively termed homology-directed repair pathways (HDR). The overarching goal of our lab is to uncover and elucidate the mechanism of homology-directed repair pathways of DNA DSBs.

Role of the DNA helicase HELQ in the maintenance of genomic stability
HELQ (PolQ-like helicase) is a 3’ to 5’ DNA helicase, which is important for cell survival when cells are exposed to genotoxic agents causing DSBs (e.g. IR and camptothecin (CPT)) and DNA inter-crosslinking lesions (ICLs, e.g. cisplatin and mitomycin C). Mice lacking functional HELQ are severely sub-fertile and more prone to developing tumours than wild-type (WT) mice. In humans, HELQ is found to be highly mutated in many cancers, especially adenomas and adenocarcinoma. Copy number variant (CNV) data also shows loss of HELQ in the majority of cancers (~74%), indicating its potential role as a tumour suppressor. Premature ovarian insufficiency (POI) is also responsible for infertility in many affected women. A major GWAS (genome-wide association study) set out to find loci associated with age at menopause and discovered the HELQ gene as one of the frequently affected genomic regions.
While HELQ’s role in DSB repair, especially during HR, has been known for more than a decade, our mechanistic understanding of its DSB repair functions has remained elusive. Only recently, we discovered that in addition to DNA unwinding activity, HELQ also possesses the capacity to proficiently anneal RPA-coated DNA strands (Figure 2 a and b). This led us to further identify its novel role in alternative DSB repair pathways, including SSA and MMEJ, and in second-end capture during HR. While these findings broadened the role of HELQ in maintaining genomic stability, they simultaneously posed interesting and important questions about the mechanistic basis of HELQ’s functions. For example, all the identified roles of HELQ in DSB repair can be solely explained by its strand annealing activity. However, HELQ is a proficient DNA helicase and how this activity contributes to HELQ’s function remains poorly understood. It is also not clear how two opposing activities, i.e. DNA annealing vs unwinding in a single enzyme, are regulated at the cellular level. We also know that RAD51 robustly stimulates HELQ’s DNA unwinding activity, but the functional significance of this stimulation in cells remains undefined. To address these interesting and important questions, our lab’s research aims to investigate both the known and novel biochemical and cellular properties of HELQ, defining its mechanistic and functional roles in maintaining genomic stability. To do so, we combine multiple approaches including biochemical analysis, single-molecule imaging (SMI) and mammalian cell-based assays in our lab.
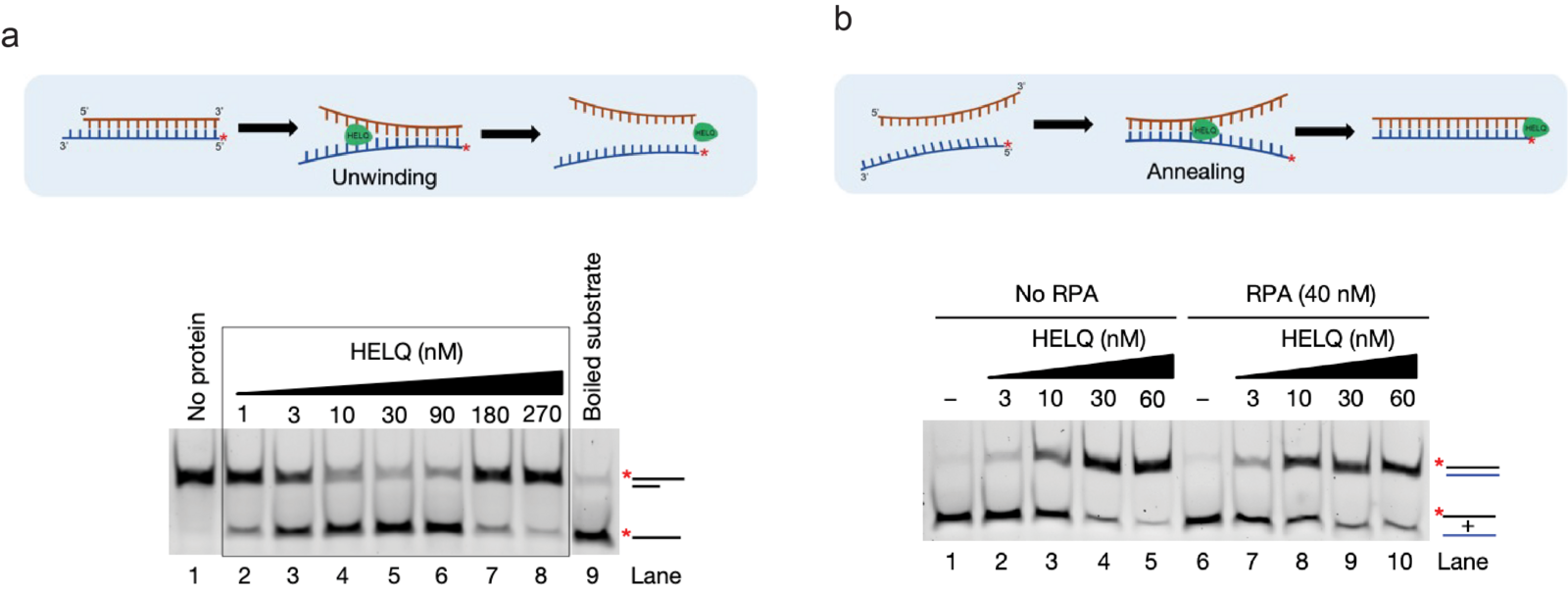
Figure 2: HELQ posseses proficient DNA unwinding and strand annealing activities. (Adapted and modified from Anand et al, Nature 2022). a) Schematics representing DNA unwinding by HELQ (top). Native polyacrylamide gel showing the unwinding of the 3’ overhang DNA substrate by HELQ (bottom). b) Schematics representing the DNA strand annealing by HELQ (top). HELQ robustly promotes annealing of complementary DNA strands, with and without RPA (bottom).
Investigation of the break-induced replication (BIR) repair pathway

HR is a predominant HDR repair pathway for repairing DSBs in cells. However, depending on the cellular context, cells can utilise alternative HDR pathways like BIR. For example, while proficient HR requires the availability of both ends of DSBs, one-ended DSBs can be repaired by BIR. These single-ended DSBs can arise specifically when a replication fork encounters pre-existing DNA lesions and collapses. Similarly, the telomeric ends of the chromosomes also represent single-ended DSBs. Mechanistically, both HR and BIR share the initial steps of DNA end resection, homology search and strand invasion (Figure 1), but the absence of a second DSB end for second-end capture results in impaired HR, and thus BIR becomes the preferred mode of DSB repair. During BIR, the invaded strand is extensively elongated by DNA synthesis inside the migrating displacement loop (D-loop), constituting the leading strand (Figure 3). This newly synthesised leading strand is displaced from behind the D-loop and used for lagging-strand formation, representing a unique conservative mode of DNA replication. In yeast, BIR can replicate the entire arm of a chromosome down to the telomeric ends.
BIR is highly mutagenic and exhibits ~1000-fold elevated mutation rates compared to normal DNA replication. This is largely due to its unusual mode of conservative replication of newly synthesised DNA, lack of the S-phase processive replisome and mismatch correction, and longer persistence of ssDNA. Moreover, microhomology-mediated BIR (MMBIR), where 1-3 base pair homologies are utilised during strand invasion, is proposed to be responsible for the gross genomic rearrangements (GCRs) found in various cancers. MMBIR also underlies GCRs found in Pelizaeus-Mezbacher disease (PMD) and MECP2 duplication syndrome, which are neurological and neurodevelopmental disorders, respectively. Interestingly, certain chromothripsis events in cancer (where massive rearrangements occur in one to a few chromosomes accompanied by copy number changes) can only be explained by MMBIR but not NHEJ, as previously suggested. In addition, BIR is hypothesised to be used in the alternative lengthening of telomeres (ALT) pathway, which occurs in 10-15% of cancers to maintain the critical length of telomeres so they can evade cell senescence/death and achieve replicative immortality. The high prevalence of ALT makes it a prime therapeutic target to treat ALT+ cancers. It should be noted that significant proportions of difficult-to-treat cancers, including osteosarcomas (63%), gliomas (30%), and neuroblastomas (24%), are ALT+. Unfortunately, these cancers are quite prevalent in children and young adults.
While yeast genetics has uncovered many features of BIR, its molecular mechanisms and the details of mammalian BIR are poorly understood. The latter is especially difficult to study due to the impressive heterogeneity and stochastic nature of BIR/ALT in mammalian cells. The research in our lab focuses on elucidating the molecular mechanisms of mammalian BIR and ALT using combined in vitro and in vivo approaches. This includes biochemical methods, SMI techniques, proteomics and cell-based fluorescent reporter assays.
If you find our research topics and area interesting, please feel free to contact us by email to find out about the available opportunities in our lab.