Dormann Lab
Research Publications Group Members BiographyMolecular mechanisms of RNA-binding protein dysfunction in neurodegenerative diseases
Our research focuses on the molecular mechanisms of neurodegenerative diseases, most notably FTD (frontotemporal dementia) and ALS (amyotrophic lateral sclerosis). FTD and ALS are currently incurable, with patients usually dying within a few years of disease onset. Existing therapies are designed to treat only the symptoms of disease and no therapies are available to slow down or stop disease progression. The main objective of our research is to obtain a molecular understanding of the mechanisms underlying FTD and ALS in order to develop new therapeutic approaches.
A common pathological hallmark of all neurodegenerative diseases is the accumulation of insoluble protein aggregates in degenerating brain areas. These protein deposits are found in neurons and glia and are intimately linked to the process of neurodegeneration. In ALS and FTD, which are clinically and genetically related diseases, the main aggregating proteins are the ubiquitously expressed RNA-binding proteins (RBPs) TDP-43 and FUS. TDP-43 aggregates occur in the majority of ALS and FTD patients, as well as in up to 50% of Alzheimer’s disease patients. In contrast, FUS aggregates are less common in ALS and FTD patients.
TDP-43 and FUS are usually found in the cell nucleus, where they regulate essential steps in mRNA processing and DNA damage repair. However, in the brain and spinal cord of ALS and FTD patients, they instead aggregate in the cytoplasm of neurons and glial cells (Figure 1). This is thought to cause neuronal dysfunction and eventually neurodegeneration through a combination of loss-of-function and gain-of-function mechanisms.

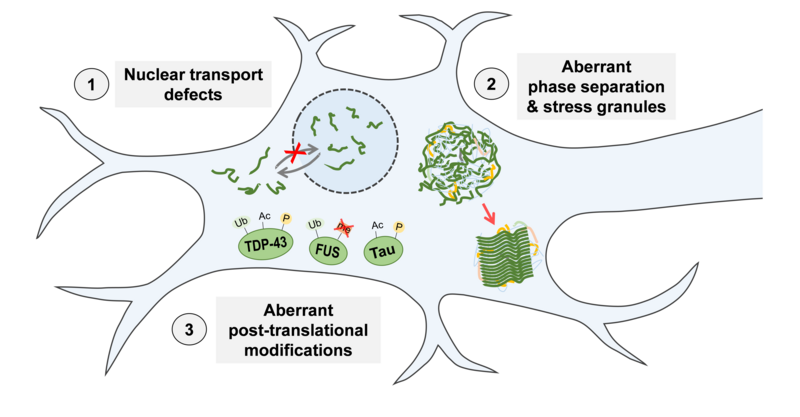
Previous research in our lab demonstrated that RBP mislocalisation and aggregation in ALS and FTD are intimately linked to molecular defects in 1) nucleocytoplasmic transport, 2) control of liquid-liquid phase separation and stress granules, and 3) post-translational modifications (PTMs, Figure 2). This is based on our finding that mutations in the nuclear localisation signal or impaired nuclear import receptor function can disrupt the import of RBPs into the nucleus in ALS and FTD patients (Dormann et al., EMBO J 2010; Hutten et al., Cell Reports 2020). The RBPs are then mislocalised to the cytosol and accumulate in stress granules, which may give rise to pathological RBP aggregates through aberrant liquid-liquid phase separation (Dormann et al., EMBO J 2010; Alberti & Dormann, Annu Rev Genetics 2019). More recently, we found that phase separation and stress granule accumulation of RBPs can be suppressed by nuclear import receptors and disease-linked PTMs, e.g. FUS arginine methylation or TDP-43 phosphorylation (Hofweber et al., Cell 2018; Gruijs da Silva et al., BioRxiv 2021). Interestingly, aberrant phase separation and disruption of nuclear transport and PTMs also appear to play a role in other neurodegenerative diseases, e.g. Huntington’s disease and Tauopathies associated with pathological aggregation of the microtubule-binding protein Tau. An important goal of our research is to understand these defects at the molecular level.
Research aims & questions
The primary aim of our research is to decipher the molecular mechanisms that underlie RBP mislocalisation and aggregation in the context of neurodegenerative diseases. We would also like to understand the physiological functions of neurodegeneration-linked RBPs, how they are regulated and how dysfunctional RBPs cause neurodegeneration. Our long-term goal is to understand the control mechanisms that maintain cellular stress resilience and protect against protein aggregation and neurodegeneration, thereby allowing healthy ageing.
We are interested in the following research questions:
(1) Role of the nuclear transport machinery in neurodegeneration & ageing:
Which cellular proteins require chaperoning by nuclear transport receptors in the cytoplasm and aggregate when transport receptors become limiting? How are the levels of nuclear transport receptors regulated in different cell types/during ageing? Can FUS and TDP-43 utilise alternative transport receptors, in addition to those that we know? How do different protein aggregates affect nucleocytoplasmic transport rates? Do they interact with and alter the nuclear pore complex?
(2) Phase separation of disease-linked proteins - functional relevance & control mechanisms:
How does phase separation affect the normal physiological functions of TDP-43 and FUS (e.g. RNA binding, RNA regulation, DNA damage repair), the dynamics of RNP granules and neuronal morphology and function? Which binding partners and PTMs control phase separation of TDP-43 and FUS? How is the cellular protein degradation machinery (proteasome, autophagy) affected by aberrant RBP phase separation, or can these systems control RBP phase separation and aggregation?
(3) Post-translational modifications of neurodegeneration-linked proteins:
Which PTMs are found on TDP-43 and FUS under normal physiological conditions, versus during cellular stress and disease? How do disease-linked PTMs on TDP-43, FUS and Tau affect their physiological localisation, function, macromolecular interactions and phase separation/aggregation behaviour? For example, how does arginine methylation regulate the RNA binding and RNA regulatory functions of FUS, and is this globally disrupted in different forms of FTD and ALS? Can we modulate disease-linked PTMs (e.g. TDP-43 phosphorylation) to counteract RBP pathology?
Experimental approach
We study the complex, multifaceted pathomechanism of ALS/FTD by dissecting it into individual components, following a reductionist approach based on in vitro reconstitution and cellular experiments. We use recombinant proteins, protein/RNA biochemistry and biophysics to understand molecular events. We employ cell lines and neuronal models to study intracellular RBP behaviour using techniques such as confocal microscopy, live cell imaging, classical biochemical and cell biological methods and proteomics. Through collaborations, we also include animal models and human post-mortem brains in our research.
Recruitment
We are expanding our team and look for motivated students and postdocs to join us! We are an international, diverse and enthusiastic research group that loves science, teamwork and a good lab spirit. Postdoc applicants are encouraged to contact Dorothee Dormann by email, including a CV, statement of research interests and the names of two referees. PhD applicants please apply through the International PhD Program, where we currently offer a PhD project.
For more information, please visit the Dormann Lab website.