Wang Lab
Research Publications Group Members BiographyTransgenerational consequences of DNA damage
One of the most fascinating facts of biology is that one organism can develop two fundamentally distinct cellular programmes: the somatic programme, which can go through development and differentiation but is destined to die, and the germline programme, which preserves a pluripotent state and is required for the perpetuation of the species. The distinction between the somatic and germline programmes is maintained by epigenetic modifications. Importantly, the somatic epigenome can be dynamically changed in response to various endogenous and exogenous stresses and affects development and ageing, while the germline epigenome remains in a repressive state to ensure a stable genome can be inherited by the consequent generations.
DNA damage poses a major threat to genome stability, chromosomal integrity and cellular function. Defects in transcription-coupled nucleotide excision repair (TC-NER) slow growth and cause mental retardation, photosensitivity and premature ageing in Cockayne syndrome (CS) patients. To ensure successful DNA repair, chromatin serves as a platform and is dynamically changed during the DNA damage response (DDR), as described by the Access-Repair-Restore model. As a crucial part of chromatin, histones are post-translationally modified via methylation, ubiquitination and acetylation to regulate DDR-related chromatin functions. Importantly, in contrast to the transient process of DNA repair, many histone modifications can leave a long-term epigenetic memory in cells and be passed down to further generations, raising the question of whether DNA damage could reshape the epigenome in damaged cells and even affect their descendants.
Due to the complexity of human epigenetics, we use C. elegans as a model to study the role of histone modifications in genome stability, longevity and transgenerational inheritance.
Long-term and transgenerational consequences of transcription-blocking DNA damage in cellular homeostasis and longevity
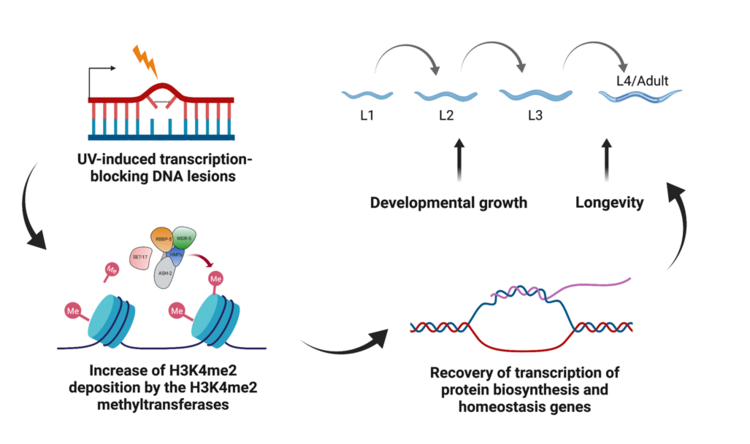
We previously found that H3K4me2 methyltransferases and demethylases in C. elegans regulate developmental growth and ageing upon UV-induced DNA damage (Figure 1). Although we only observed a temporary increase in H3K4me2 upon UV irradiation, prolonged lifespan in C. elegans after UV damage indicates that these histone modification changes may leave a long-term beneficial effect on the animals that recover from DNA damage. Importantly, H3K4 methylation is also widely studied for its role in transgenerational epigenetic inheritance, a highly conserved phenomenon observed in both plants and animals. In C. elegans, mutations in the MLL/COMPASS complex and SPR-5 caused a heritable increase in longevity for multiple generations. These lines of evidence indicate that the deposition of H3K4me2 in parents might not be completely reset between generations. We therefore hypothesise that DNA damage-induced H3K4me2 might cause a long-term and transgenerational effect on longevity in the offspring.
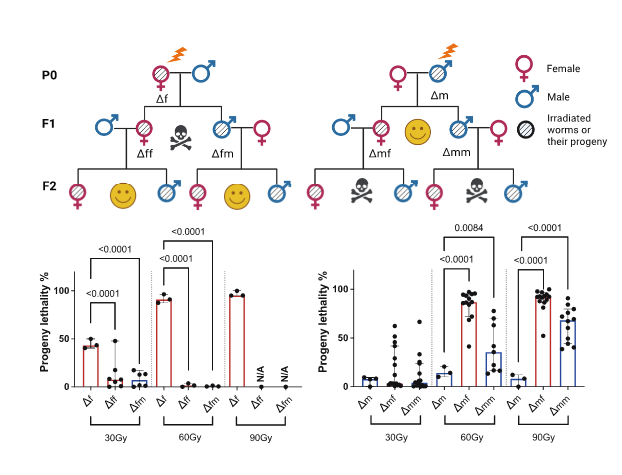
Transgenerational epigenetic “memory” of paternal ionising radiation (IR) exposure
Previous studies in mammals have demonstrated that paternal exposure to a variety of environmental factors affects the risk of disease in subsequent generations, although the mechanism behind this phenomenon is still unclear. In C. elegans, we observed that DNA double-strand breaks (DSBs) in mature sperm led to high embryonic lethality in the F2 generation (Figure 2). This transgenerational lethality is attributed to the persistence of DNA DSBs in the F1 generation, where a highly enriched heterochromatin structure blocks the accessibility of homologous recombination (HR) repair machinery. The main questions I address here are how DNA damage in sperm alters the epigenome of the offspring and whether these changes in the epigenome can affect genome stability and longevity in subsequent generations.
Transgenerational effect of paternal reproductive ageing and chronic radiation exposure
In humans, nearly 80% of de novo mutations originate from the paternal germline; moreover, the germline mutation rate is highly correlated with paternal age. Indeed, advanced paternal age is associated with various congenital diseases, schizophrenia, autism and reproductive defects. By analysing the public human genome dataset, we made a striking observation that paternal de novo mutations in humans display a structural change similar to the change we detected in worms, suggesting the mechanism we discovered may be conserved across species and contribute to genome diversification during evolution. This observation encouraged me to use C. elegans to investigate the transgenerational effect of DNA damage from paternal reproductive ageing. Moreover, humans can be exposed to various sources of radiation as they age and these radiation sources are usually from natural backgrounds or specific working environments, such as nuclear power plants. This long-term, low-dose radiation exposure is defined as chronic radiation exposure and can contribute to the elevated mutation rate in the paternal germline, due to the lack of efficient repair machinery in sperm. It is therefore crucial to understand if chronic radiation exposure also leads to a transgenerational effect. To this end, I will examine the genetic and epigenetic transgenerational effect of paternal reproductive ageing and chronic radiation exposure using C. elegans as a model.
The distinction between maternal and paternal DNA repair mechanisms during embryogenesis
In our previous work, we found that after fertilisation maternal DNA damage can be either accurately repaired by HRR or eliminated via embryonic mitotic catastrophe without being transmitted to subsequent generations. On the other hand, paternal DNA damage can only engage the maternally-provided error-prone DNA repair machinery TMEJ. Therefore, what triggers the different choices of DNA repair between maternal and paternal genomes after fertilisation is a fascinating question. I hypothesise that the paternal genome decondensation process following fertilisation may affect the choice of DNA repair mechanism, as the highly compacted chromatin is normally considered a barrier to HRR engagement. To this end, I will use immunofluorescence or FISH approaches to examine chromatin conformation changes in the paternal and maternal genomes after fertilisation. I will track the localisation of different DNA repair proteins in response to chromatin remodelling. Subsequently, depleting regulators involved in chromatin decondensation might improve the DNA repair fidelity of the paternal genome. Taken together, this work will provide insight into the molecular pathways governing paternal genome stability and might identify potential therapeutic targets for paternally-inherited congenital diseases.