Ulrich Lab
Research Publications Group Members Biography Team Leader - Reuter Team Leader - WollscheidUbiquitin, SUMO & the maintenance of genome stability
Our lab studies the regulatory mechanisms that contribute to ensuring the complete and accurate duplication of a cell’s genetic information in every cell cycle, especially in the face of DNA damage. We are particularly interested in how posttranslational protein modifiers of the ubiquitin family, such as ubiquitin and SUMO, control genome maintenance pathways, and how they employ their versatile signalling capacity to regulate other cellular processes.
Regulation of DNA damage bypass in time & space
Dealing with damage during DNA replication is particularly important because the replication machinery cannot cope with defective templates. In order to avoid a permanent arrest in this situation, cells have developed mechanisms of damage bypass. In contrast to most DNA repair systems, which usually restore the original sequence information, DNA damage bypass allows the replication machinery to “skip over” lesions without their actual removal. It ensures the completion of DNA replication on damaged templates and is therefore essential for the survival of a cell in the presence of genotoxic stress. As lesion bypass is often associated with damage-induced mutations, however, the pathway is also a potential source of genome instability in itself and therefore needs to be tightly controlled.
We found that in budding yeast, most lesions in the replication template do not cause replication fork stalling, but instead lead to the emergence of postreplicative daughter-strand gaps by polymerase re-priming. Both checkpoint signalling and lesion bypass are then initiated within these single-stranded regions, following their expansion by an exonuclease. The sites of damage bypass are distinct from the locations of double-strand break repair. Instead, they form within replicated DNA in regions that we now call “POstreplicative Repair Territories”, or PORTs (Figure 1).
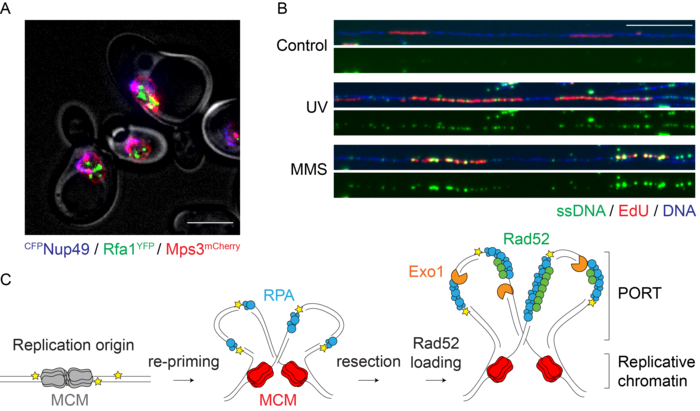
Figure 1. A. Sites of DNA damage bypass (Rfa1GFP foci, green) are largely distinct from nuclear pores (CFPNup49, blue) and the nuclear periphery (Mps3mCherry, red). Scale bar: 5 µm. B. DNA fibre analysis shows daughter-strand gaps (ssDNA, green) emerging in newly synthesised DNA (EdU, red) after UV irradiation or MMS treatment. Scale bar: 20 kbp. C. Model for the formation of PORTs by polymerase re-priming and daughter-strand gap expansion.
Our current research focuses on the following questions:
- What is the response of the replisome to polymerase-blocking lesions?
- What is the contribution of chromatin to DNA damage bypass?
- What determines the accuracy of DNA damage bypass?
In order to address these questions, we use microscopic imaging of fluorescently tagged repair proteins, replication analysis by means of DNA combing, as well as designed fluorescent in vivo sensors of DNA damage bypass. Moreover, we have developed a system to introduce small DNA lesions locally into defined regions of the genome, allowing us to use chromatin immunoprecipitation combined with quantitative PCR or next-generation sequencing to monitor the passage of the replisome and the recruitment of relevant repair factors in a time-resolved manner with high spatial resolution.
Ubiquitin & SUMO signalling in DNA damage bypass & beyond
Ubiquitin and SUMO are small posttranslational modifiers involved in almost all signalling pathways of eukaryotic cells. When covalently coupled to other proteins, they modulate their interactions, localisation or stability. The striking versatility of ubiquitin comes from its ability to form polymeric chains of varying geometries, which are thought to convey distinct biological signals through interactions with downstream effector proteins harbouring dedicated ubiquitin-binding domains.
In the context of DNA damage bypass, ubiquitin and SUMO control the properties of the essential replication factor, PCNA. Whereas monoubiquitylation activates translesion synthesis by damage-tolerant DNA polymerases, polyubiquitylation via lysine (K) 63-linked polyubiquitin chains is required for an error-free pathway of damage avoidance involving template switching. In budding yeast, PCNA is also sumoylated during DNA replication, which prevents unwanted recombination events and allows the ubiquitin-dependent reactions to proceed in response to replication stress. Whereas the consequences of PCNA monoubiquitylation and sumoylation are comparatively well understood, little is known about the mechanism by which K63-polyubiquitylation activates template switching.
We are investigating the signalling function of polyubiquitin chains in relation to PCNA as well as other biological processes, focussing on the following questions:
- What are the downstream effectors of K63-polyubiquitylated PCNA?
- What is the relevance of polyubiquitin chain linkage for ubiquitin writers and readers?
- How are the dynamics of ubiquitin and SUMO conjugation regulated in cells?
We are currently generating tools and methods to monitor and manipulate ubiquitin and SUMO in budding yeast and vertebrate cells to address these questions (Figure 2). These include specific probes based on nanobodies and synthetic scaffolds such as Designed Ankyrin Repeat Proteins (DARPins). In addition, we are developing tailor-made ubiquitin ligases and matching ubiquitin acceptor sites for the inducible, substrate- and linkage-specific polyubiquitylation of any desired target protein. This approach, which we named “Ubiquiton” in analogy to the well-known “Degron” approach, will allow us to investigate ubiquitin signalling by direct re-programming of polyubiquitin chain linkage.
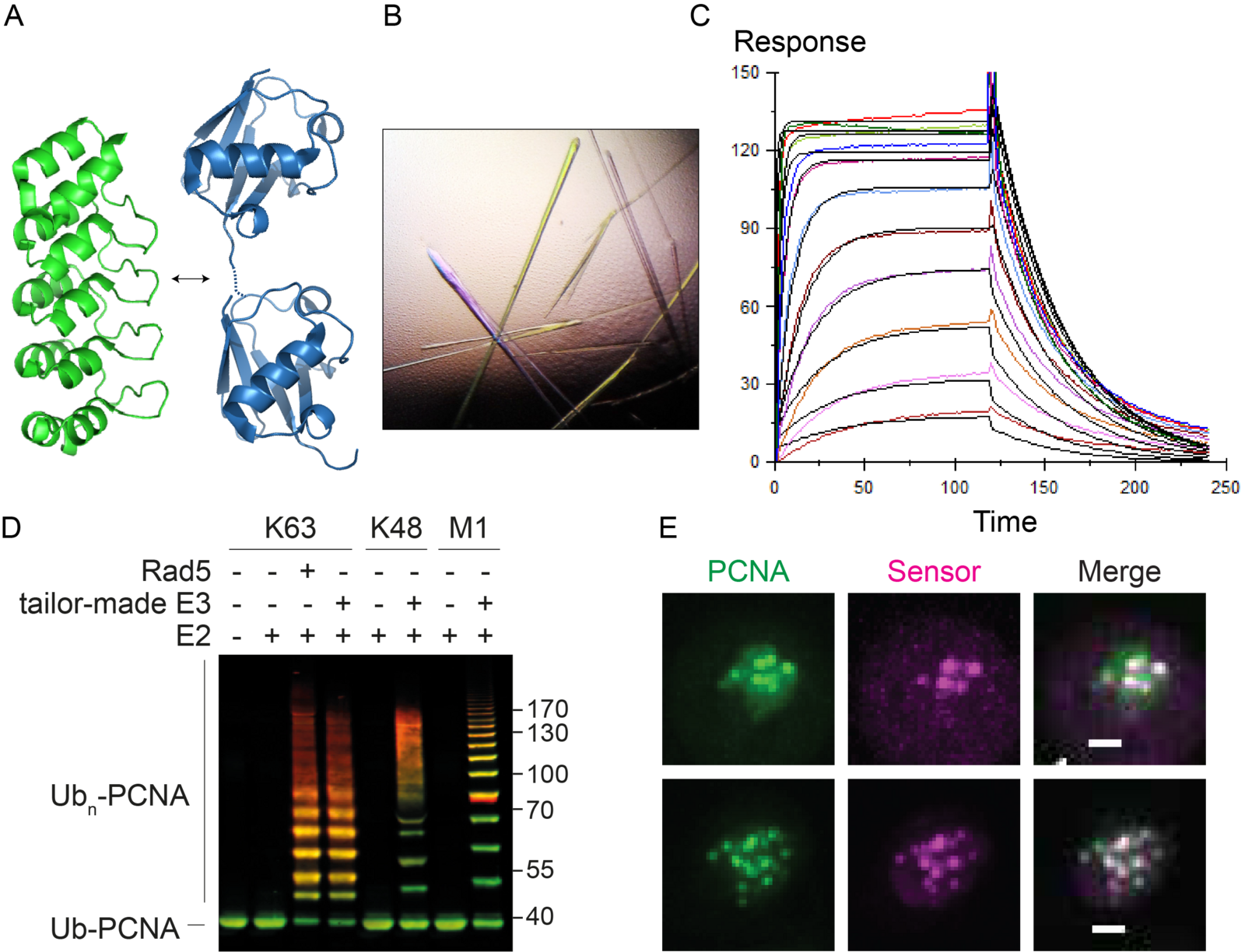
Figure 2. A. The DARPin scaffold (PDB 4J7W, green) as a basis for linkage-selective ubiquitin probes (PDB 5GOJ, blue). B. Crystals of a complex of yeast SUMO and a selective DARPin. C. Surface plasmon resonance analysis of a DARPin-SUMO complex. D. In vitro polyubiquitylation of PCNA with tailor-made enzymes of distinct linkage selectivities, detected by western blotting (green: PCNA; red: ubiquitin). E. Co-localisation of GFPPCNA (green) and a fluorescent probe selective for ubiquitylated PCNA (magenta) in live yeast cells in response to DNA damage. Scale bar: 1 µm.
Ubiquitin-dependent genome maintenance in mammalian cells
Given the greater complexity of their signalling pathways, mammalian cells present an even larger variety of regulatory processes involving ubiquitin and/or SUMO than budding yeast. Our lab is therefore investigating selected aspects of the DNA damage response unique to higher eukaryotes. In this context, we are studying the mammalian ubiquitin conjugation enzymes acting on PCNA in damage bypass, but also factors generally known for their contribution to transcriptional regulation and intracellular motility, such as the ubiquitin-binding motor protein Myosin VI.
Genetic manipulations via CRISPR/Cas9-mediated genome editing, lentiviral transduction and classical transfections of overexpression constructs or siRNAs in standard human cell lines allow us to investigate the roles of these proteins in genome maintenance. To this end, we have established a wide range of functional assays, including survival and cell cycle analysis, DNA combing, resection and traffic light reporter assays as well as live-cell fluorescence microscopy and immunofluorescence analysis of the DNA damage response (Figure 3).

Figure 3. A. The actin cytoskeleton of A549 cells is monitored by phalloidin staining (red), the DNA by DAPI (white). B. Metaphase spreads detect chromosome abnormalities. C. Flow cytometry analysis monitors damage signalling (γH2AX) and apoptosis (cleaved PARP-1). D. Defects in the replication stress response cause anaphase bridges during cell division (blue: DNA; green: PICH; red: FANCD2). E. Staining for γH2AX (green) and 53BP1 (red) reveals the structure of damage-induced nuclear bodies.
Tools for the genome-wide analysis of DNA replication & repair
Genome-wide methods for the mapping of lesions and replication intermediates have become important tools for genome stability research. Analysis of daughter-strand gaps and stalled replication forks in this manner would require the detection of DNA termini not associated with double-strand breaks. Yet, single-strand breaks have so far eluded systematic analysis by next-generation sequencing. We have now developed a sequencing technique based on the “Genome-wide Ligation of 3’-OH DNA Ends” (GLOE-Seq) for the mapping of such structures with nucleotide resolution (Figure 4). Beyond single-strand breaks, our method is also applicable to other lesions that can be converted to single-strand breaks enzymatically, thus expanding the range of potential applications.
After validating the method on in vitro-digested DNA, we have begun to explore the power of GLOE-Seq in proof-of-principle experiments addressing the distribution of single-strand breaks in unperturbed as well as ligase-deficient cells. We have also mapped DNA lesions and repair intermediates after exposure to UV irradiation and alkylating agents. Our analysis has revealed surprising biases and asymmetries of spontaneous single-strand breaks, distinct from the patterns of DNA double-strand breaks, and it has shown that GLOE-Seq is suitable for the mapping of Okazaki fragments without prior size selection in yeast and human cells. We are currently refining the method in order to further expand its scope, and we are using GLOE-Seq to gain insight into the impact of DNA-damaging agents on DNA replication patterns and damage bypass activity.
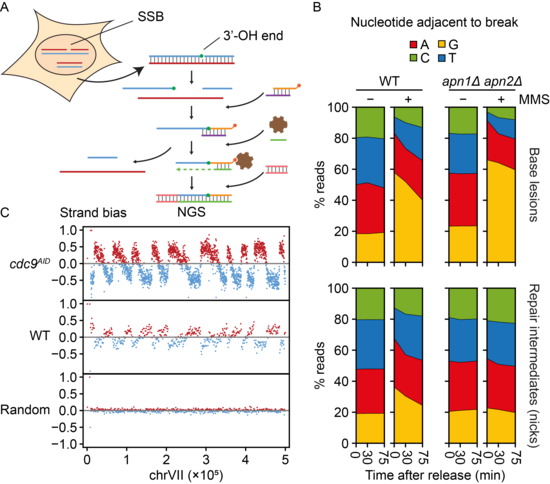
Figure 4. A. GLOE-Seq workflow. B. Mapping of base lesions and repair intermediates (single-strand breaks) in wildtype (WT) and base excision repair-deficient yeast (apn1Δ apn2Δ) by GLOE-Seq serves to analyse DNA repair kinetics. C. GLOE-Seq allows the mapping of Okazaki fragments and replication fork direction in ligase-depleted cells (cdc9AID) and reveals a complementary strand bias of nicks in WT cells.