Beli Lab
Research Publications Group Members BiographyChromatin biology & proteomics
Genome stability maintenance
Maintenance of genome stability is essential for the prevention of cancer and premature aging. A complex network of proteins and signalling pathways ensures genome maintenance in response to external stressors such as UV light as well as in response to DNA damage introduced through physiological processes such as DNA replication and transcription. The research in our group focuses on identifying and understanding the mechanisms that function in the cellular DNA damage response. To achieve these goals, we are developing and employing mass spectrometry-based proteomics methods to analyse DNA damage-dependent protein-protein interactions as well as phosphorylation and ubiquitin-dependent mechanisms that regulate the DNA damage response. Ongoing projects in the lab aim to gain insights into how transcription and protein complexes involved in transcription initiation and elongation are regulated in response to transcription-blocking lesions introduced by UV light. Furthermore, we are investigating the protein-based mechanisms that function to regulate the levels of R-loops that frequently form during transcription with the goal to better understand the contribution of R-loop dysregulation to disease.
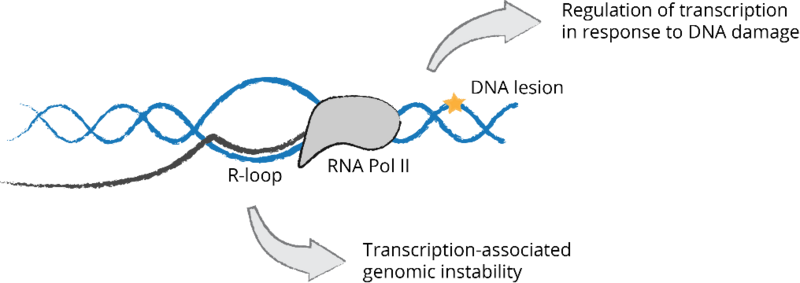
Interplay between transcription and genome stability maintenance. Transcription can pose a threat to genome stability through the frequent formation of RNA-DNA hybrids with a displaced single-stranded DNA or R-loops. Transcription-blocking lesions induced by UV light trigger global regulation of different RNA metabolic processes including transcription, alternative splicing and RNA stability.
Ubiquitin signalling in proteome quality control
During gene expression, quality control pathways monitor each step to detect aberrant mRNAs and proteins. These mechanisms ensure protein homeostasis and are essential to prevent neurodegenerative diseases. The research in our group aims to identify and understand ubiquitin-dependent mechanisms that regulate proteome quality control and dynamics. To this end, we employ quantitative ubiquitin remnant profiling to obtain a global view on ubiquitylation events that regulate stress-induced protein quality control pathways. Furthermore, we develop and employ mass spectrometry-based proteomics approaches to better understand the cellular function and substrates of atypical ubiquitin chains such as K6-linked ubiquitylation. We also study the contribution of ubiquitin-proteasome system and autophagy to targeted protein degradation in different cellular compartments.
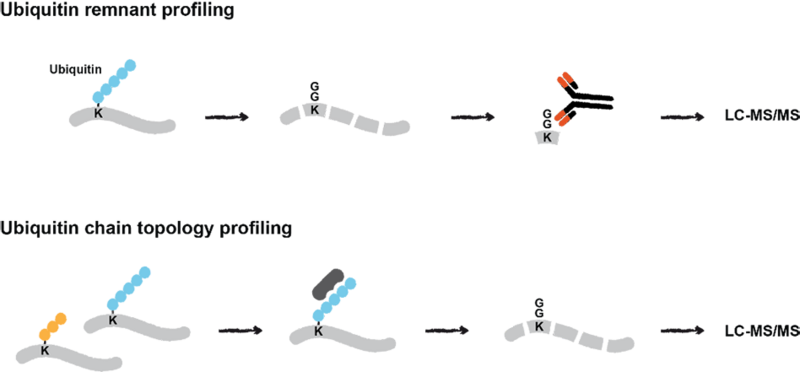
Quantitative mass spectrometry-based proteomics approaches for interrogation of ubiquitin signalling. Ubiquitin remnant profiling enables quantification of endogenous ubiquitylation sites in response to cellular stress. The presence and abundance of different ubiquitin chains on proteins is investigated by their enrichment using chain-specific binders and subsequent LC-MS/MS analysis.
To approach the research questions that focus on these topics, we develop and employ mass spectrometry-based approaches for quantitative analysis of protein-protein interactions as well as protein phosphorylation and ubiquitylation. We combine quantitative proteomics with a range of methodologies including high content microscopy, genomics, functional DNA repair assays as well as classical biochemical and cell biological methods.
Funding
The group is part of different national and international research consortia focusing on genome stability maintenance, ubiquitin signalling and RNA biology, including the Collaborative Research Center SFB 1361 on Regulation of DNA Repair and Genome Stability, Collaborative Research Center SFB 1551 on Polymer Concepts in Cellular Function, Collaborative Research Center SFB 1678 on Systemic Consequences of Fidelity Changes in mRNA and Protein Biosynthesis, Collaborative Research Center SFB 1678, Collaborative Research Center TRR 387 UbiQancer - Functionalizing the Ubiquitin System against Cancer, Research Unit 2800 on Chromosome Instability, and Research Training Group 4R on R-loop Regulation in Robustness and Resilience.
Recruiting
We are always searching for motivated postdoctoral fellows, PhD students and Master/Bachelor students to join our team. Postdoc applicants are encouraged to contact Petra Beli directly by email including a CV, statement of research interest and/or project idea. PhD applicants please apply through the International PhD Program. We also welcome applications from computational scientists interested in projects involving the analysis and integration of quantitative proteomics data.